Testing support for the northern and southern dispersal routes out of Africa: an analysis of Levantine and southern Arabian populations Authors Deven N. Vyas, Ali Al-Meeri, Connie J. Mulligan First published: 15 September 2017
Abstract
Objectives
The Northern Dispersal Route (NDR) and Southern Dispersal Route (SDR) are hypothesized to have been used by modern humans in the dispersal out of Africa. The NDR follows the Nile into Northeast Africa and crosses the Red Sea into the Levant. The SDR emerges from the Horn of Africa and crosses the Bab el-Mandeb into southern Arabia. In this study, we analyze genetic data from populations living along the NDR and SDR to test support for each dispersal route. Materials and methods
We genotyped 90 Yemeni samples on the Affymetrix Human Origins array. We analyzed these data with published data from Levantine and other southern Arabian populations as well as 157 comparative populations for a total sample size of >550,000 genetic variants from >2,000 individuals in >160 populations. We calculated outgroup f3 statistics to test how Levantine and southern Arabian populations relate to African populations living along the NDR and SDR and to other non-African populations. Results
We find that Levantine and southern Arabian populations bear similar genetic relationships to both African and non-African populations, thus providing no support for the use of one dispersal route over the other. Discussion
Our results are consistent with a history of gene flow between the Levant and southern Arabia. Consideration of genetic, archaeological, and paleoclimate data provide a slight edge for the SDR but, ultimately, more data are needed to definitively identify which dispersal route out of Africa was used.
7-1-2016 The multiple histories of Western Asia: Perspectives from ancient, and modern genomes Recep Taskent University at Buffalo, Omer Gokcumen University at Buffalo,
Abstract: Anthropological genetics has revolutionized the way we study variation in human populations, their relationships with each other and with past populations. Since the very early days of the discipline, Western Asia has been a major focus (Menozzi et al. 1978). After all, it is the geographical focal point where Africa, Asia, and Europe meet and it is the hotbed of cultural innovation, most notably the emergence of settled Neolithic communities (Gordon Childe 1936; Mellaart 1967; Barker 2009). As such, it has been central to most major Eurasian civilizations (Kuhrt 1995; Gregory 2010), and, more recently, a dynamic mix of tribal and ethnic units, religious sects, and national identities. Some questions emerge as central within the broader framework of Western Asian genetic variation: Who are the ancestors of Western Asian populations? How did contemporary and ancient Western Asians contribute to the peopling of the rest of Eurasia? Which routes in Western Asia did the first migrants out of Africa take? Who were the first farmers? Where, when, and to what extent did Neanderthals contribute to the gene pools of Eurasian ancestors? In this paper, we review the latest genetics research tackling these questions, with special emphasis on the recently available ancient genomics datasets, as well as the emerging notion that ancient interactions among human populations are more important than previously thought.
Parallel ancient genomic transects reveal complex population history of early European farmers Wolfgang Haak, David Reich doi: https://doi.org/10.1101/114488
Abstract
Ancient DNA studies have established that European Neolithic populations were descended from Anatolian migrants who received a limited amount of admixture from resident hunter-gatherers. Many open questions remain, however, about the spatial and temporal dynamics of population interactions and admixture during the Neolithic period. Using the highest-resolution genome-wide ancient DNA data set assembled to date---a total of 177 samples, 127 newly reported here, from the Neolithic and Chalcolithic of Hungary (6000-2900 BCE, n = 98), Germany (5500-3000 BCE, n = 42), and Spain (5500-2200 BCE, n = 37)---we investigate the population dynamics of Neolithization across Europe. We find that genetic diversity was shaped predominantly by local processes, with varied sources and proportions of hunter-gatherer ancestry among the three regions and through time. Admixture between groups with different ancestry profiles was pervasive and resulted in observable population transformation across almost all cultural transitions. Our results shed new light on the ways that gene flow reshaped European populations throughout the Neolithic period and demonstrate the potential of time-series-based sampling and modeling approaches to elucidate multiple dimensions of historical population interactions. ________________________________________________
May 2017, Volume 136, Issue 5, pp 529546 | Cite as
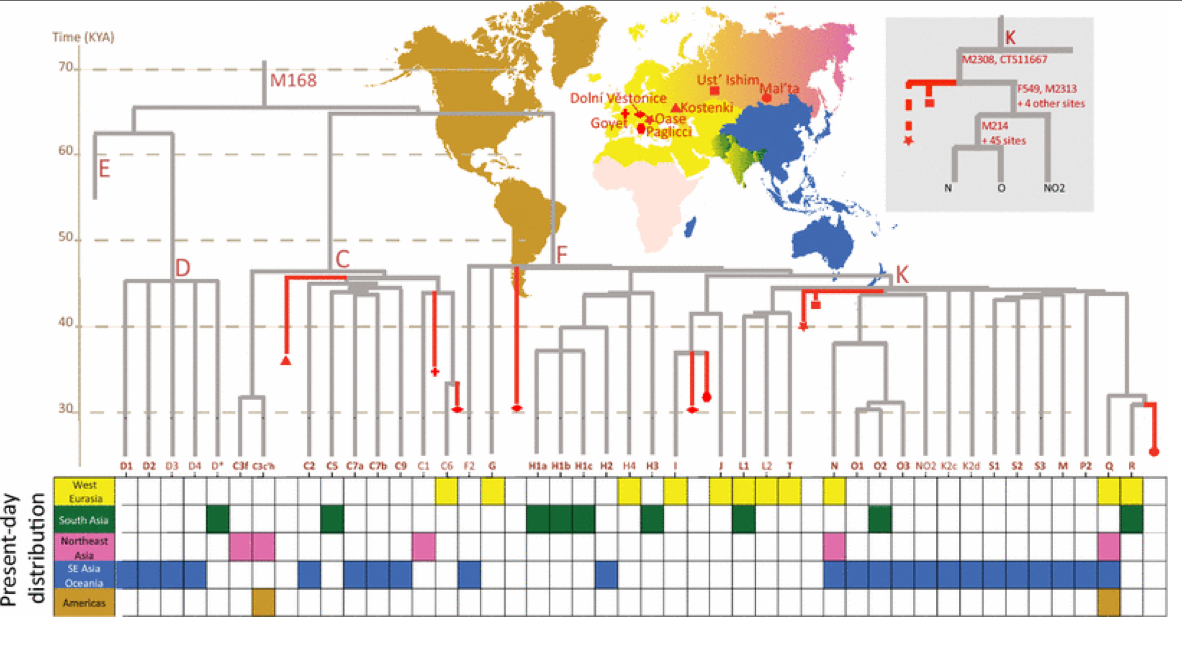
The study of human Y chromosome variation through ancient DNA
Abstract
High throughput sequencing methods have completely transformed the study of human Y chromosome variation by offering a genome-scale view on genetic variation retrieved from ancient human remains in context of a growing number of high coverage whole Y chromosome sequence data from living populations from across the world. The ancient Y chromosome sequences are providing us the first exciting glimpses into the past variation of male-specific compartment of the genome and the opportunity to evaluate models based on previously made inferences from patterns of genetic variation in living populations. Analyses of the ancient Y chromosome sequences are challenging not only because of issues generally related to ancient DNA work, such as DNA damage-induced mutations and low content of endogenous DNA in most human remains, but also because of specific properties of the Y chromosome, such as its highly repetitive nature and high homology with the X chromosome. Shotgun sequencing of uniquely mapping regions of the Y chromosomes to sufficiently high coverage is still challenging and costly in poorly preserved samples. To increase the coverage of specific target SNPs capture-based methods have been developed and used in recent years to generate Y chromosome sequence data from hundreds of prehistoric skeletal remains. Besides the prospects of testing directly as how much genetic change in a given time period has accompanied changes in material culture the sequencing of ancient Y chromosomes allows us also to better understand the rate at which mutations accumulate and get fixed over time. This review considers genome-scale evidence on ancient Y chromosome diversity that has recently started to accumulate in geographic areas favourable to DNA preservation. More specifically the review focuses on examples of regional continuity and change of the Y chromosome haplogroups in North Eurasia and in the New World.
An open question in human evolution is the importance of polygenic adaptation: adaptive changes in the mean of a multifactorial trait due to shifts in allele frequencies across many loci. In recent years, several methods have been developed to detect polygenic adaptation using loci identified in genome-wide association studies (GWAS). Though powerful, these methods suffer from limited interpretability: they can detect which sets of populations have evidence for polygenic adaptation, but are unable to reveal where in the history of multiple populations these processes occurred. To address this, we created a method to detect polygenic adaptation in an admixture graph, which is a representation of the historical divergences and admixture events relating different populations through time. We developed a Markov chain Monte Carlo (MCMC) algorithm to infer branch-specific parameters reflecting the strength of selection in each branch of a graph. Additionally, we developed a set of summary statistics that are fast to compute and can indicate which branches are most likely to have experienced polygenic adaptation. We show via simulations that this method - which we call PhenoGraph - has good power to detect polygenic adaptation, and applied it to human population genomic data from around the world. We also provide evidence that variants associated with several traits, including height, educational attainment, and self-reported unibrow, have been influenced by polygenic adaptation in different human populations.
Posts: 42922 | From: , | Registered: Jan 2010
| IP: Logged |




 UBBFriend: Email this page to someone!
UBBFriend: Email this page to someone!





 Printer-friendly view of this topic
Printer-friendly view of this topic