Using STR-FM to assess error profiles in the context of STRs for Illumina data Detecting STRs in short reads
Abstract Go to: Background Short tandem repeats (STRs) are found in many prokaryotic and eukaryotic genomes, and are commonly used as genetic markers, in particular for identity and parental testing in DNA forensics. The unstable expansion of some STRs was associated with various genetic disorders (e.g., the Huntington disease), and thus was used in genetic testing for screening individuals at high risk. Traditional STR analyses were based on the PCR amplification of STR loci followed by gel electrophoresis. With the availability of massive whole genome sequencing data, it becomes practical to mine STR profiles in silico from genome sequences. Software tools such as lobSTR and STR-FM have been developed to address these demands, which are, however, built upon whole genome reads mapping tools, and thus may not be sensitive enough.
Go to: Results In this paper, we present a standalone software tool STRScan that uses a greedy algorithm for targeted STR profiling in next-generation sequencing (NGS) data. STRScan was tested on the whole genome sequencing data from Venter genome sequencing and 1000 Genomes Project. The results showed that STRScan can profile 20% more STRs in the target set that are missed by lobSTR.
Go to: Conclusion STRScan is particularly useful for the NGS-based targeted STR profiling, e.g., in genetic and human identity testing. STRScan is available as open-source software at http://darwin.informatics.indiana.edu/str/.
Abstract Short tandem repeats, specifically microsatellites, are widely used genetic markers, associated with human genetic diseases, and play an important role in various regulatory mechanisms and evolution. Despite their importance, much is yet unknown about their mutational dynamics. The increasing availability of genome data has led to several in silico studies of microsatellite evolution which have produced a vast range of algorithms and software for tandem repeat detection. Documentation of these tools is often sparse, or provided in a format that is impenetrable to most biologists without informatics background. This article introduces the major concepts behind repeat detecting software essential for informed tool selection. We reflect on issues such as parameter settings and program bias, as well as redundancy filtering and efficiency using examples from the currently available range of programs, to provide an integrated comparison and practical guide to microsatellite detecting programs.
xyyman Member # 13597
posted
What do you all think?
You will need Download the lobSTR package. Download the lobSTR resource bundle for the appropriate reference genome (available on the downloads page). If you'd like to use your own custom set of STR loci, see Building a custom reference before proceeding. Announcements As of v4+, allelotype can now take input BAM files generated by aligners other than lobSTR. The only aligner that has been tested and shown to work well is BWA-MEM. For more details on running allelotype with BWA-MEM-generated BAMs as input, see Best practices for using lobSTR with BWA-MEM alignments Using noise models generated by previous allelotype versions (before v3.0.0) with versions 3.0.0 and above will result in erroneous genotype calls. If using allelotype 3.0.0+, you must use the new noise model (illumina.v3.pcrfree) or a custom noise model built with the
Basic usage (as of v4.0.0) To run lobSTR using default parameters: Single-end reads:
posted
He can use lobstr to pull str reads from bam files.... it's the easiest way to genotype STrs.
IDK about that STRscan stuff... I need to see it's results. Haven't used it.
Also I don't see where we can get STR's from SNPs... What are you referring to Xyyman?
xyyman Member # 13597
posted
Ok I will make it simple. Since you are playing dumb. The Abusir data are in BAM files. Can you pull STR from BAM files using any of these software? It looks like it is possible. Thoughts?
Elmaestro Member # 22566
posted
NOOOOOOOOOOOOOOOOOOOOOO
If you really care to help in the way you think you are, you should at-least try to understand what you're talking about.
I have to admit...I still need to get this straight in my head
xyyman Member # 13597
posted
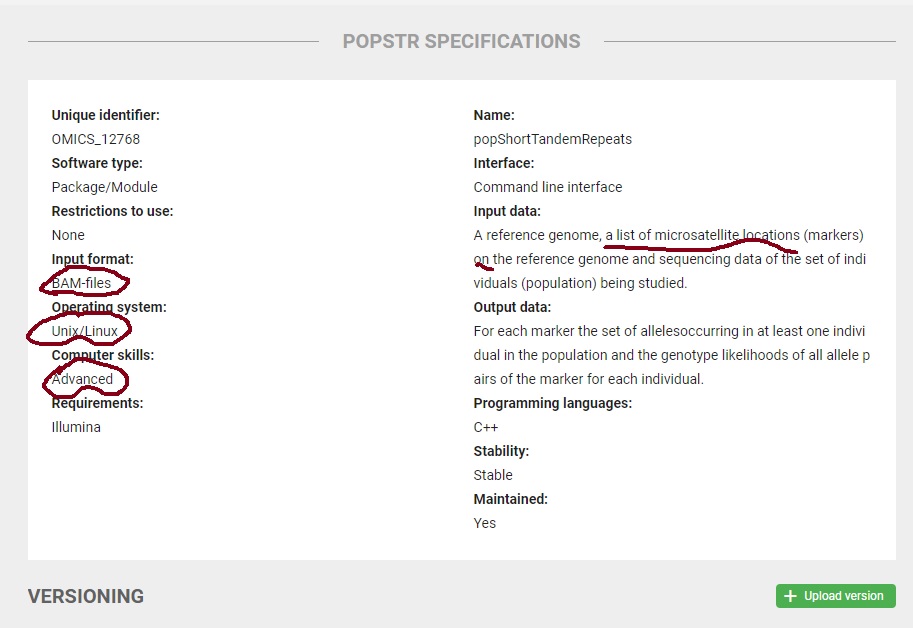
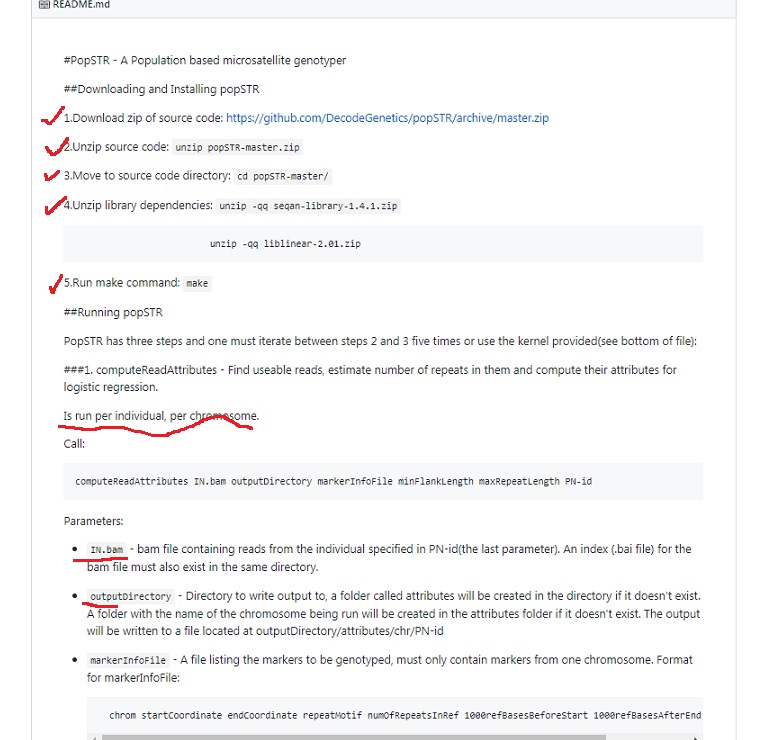
popSTR: population-scale detection of STR variants. Kristmundsdóttir S1, Sigurpálsdóttir BD2, Kehr B1, Halldórsson BV1,2. Author information
Abstract MOTIVATION: Microsatellites, also known as short tandem repeats (STRs), are tracts of repetitive DNA sequences containing motifs ranging from two to six bases. Microsatellites are one of the most abundant type of variation in the human genome, after single nucleotide polymorphisms (SNPs) and Indels. Microsatellite analysis has a wide range of applications, including medical genetics, forensics and construction of genetic genealogy. However, microsatellite variations are rarely considered in whole-genome sequencing studies, in large due to a lack of tools capable of analyzing them.
RESULTS: Here we present a microsatellite genotyper, optimized for Illumina WGS data, which is both faster and more accurate than other methods previously presented. There are two main ingredients to our improvements. First we reduce the amount of sequencing data necessary for creating microsatellite profiles by using previously aligned sequencing data. Second, we use population information to train microsatellite and individual specific error profiles. By comparing our genotyping results to genotypes generated by capillary electrophoresis we show that our error rates are 50% lower than those of lobSTR, another program specifically developed to determine microsatellite genotypes.
The latest versions of samtools have the bam2fq command. Chain that to seqtk seq like so:
samtools bam2fq input.bam | seqtk seq -A > output.fa
For future record: samtools versions >1.3 can convert bam to fasta directly via samtools fasta
samtools --help
Program: samtools (Tools for alignments in the SAM format) Version: 1.4 (using htslib 1.4)
Usage: samtools <command> [options]
Commands: [...] -- File operations [...] fastq converts a BAM to a FASTQ fasta converts a BAM to a FASTA
It can be done. STR from GWAS??!!
xyyman Member # 13597
posted
Overview One of the most common questions we receive is about how to use lobSTR to genotype Y-STRs and the CODIS set from sequencing data. Both are useful for identification in forensics settings, and Y-STRs are also used both in the genetic genealogy community and population genetics. Here we give a short tutorial on how to go from the lobSTR VCF output to a list of Y-STR and CODIS genotypes in standard nomenclature. As an example we will use the Venter genome.
quote:Originally posted by Elmaestro: Bad news, after multiple attempts and methods I failed to get any useful STR data... Even from the fastq files. All the STR calls I got were generic. I couldn't Identify them with any program so I even checked what was going on manually (by literally looking at the loci), and it turns out their's no bps at all in the regions where we'd find these markers... so to answer Beyokus Question above no, neither do the Fastq files have em, they probably weren't even sequenced.
If anyone want a copy of the generic STR calls for whatever reason pm me, though I might warn you, you're on your own from there... I haven't bothered classifying them or researching the nomenclature.

 posted
posted