So, did these "single individuals" had admixture, or was "purely" these SNPs found in these "single individuals"? Considering that most seem to carry subclades of some sort.
quote:Originally posted by the lioness,: why are you saying they are low in frequency?
Because it is, hence my previous statement.
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
quote:Originally posted by Ish Gebor: @ the lioness, how do you feel about the nazi history these institutes have? I am referring to University of Tuebingen and Max Planck.
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
I'm not really sure how to read this. Which numbers/columns are we suppose to be reading to figure out which group they belonged to.
Posts: 2508 | From: . | Registered: Nov 2011
| IP: Logged |
posted
To those who can follow. Here is what I think how we can process the Abusir mummies.
1. Download the free BAM files of the 92 Abusir mummies. 2. Use IGV tools to process each individual. Convert BAM(binary) to SAM(text delimited) 3. Download or upload files to use ALFRED to ID the SNPs strings or STRs. Eg TPOX is TPOX (AATG) n tetranucleotide repeat,
Thoughts anyone?
Also we can download TreeMix and process in this software. Lot of work? I understand TreeMIx only runs on Mac or Ubuntu? Anyone?
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
I want to add. Anyone has the address for the pop STRs eg for TPOX? rsXXXXXX?
quote:Originally posted by xyyman: To those who can follow. Here is what I think how we can process the Abusir mummies.
1. Download the free BAM files of the 92 Abusir mummies. 2. Use IGV tools to process each individual. Convert BAM(binary) to SAM(text delimited) 3. Download or upload files to use ALFRED to ID the SNPs strings or STRs. Eg TPOX is TPOX (AATG) n tetranucleotide repeat,
Thoughts anyone?
Also we can download TreeMix and process in this software. Lot of work? I understand TreeMIx only runs on Mac or Ubuntu? Anyone?
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
Anyone can jump in any time....(wink). Capra, TheMaster, AstenB
We do not have Lucas Martin and DNATribes to show us how it is done any more. But we have more freeware and more sophisticated tools today. What about this?
Download Abusir mummies genome which are BAM Files. Convert the BAM file to FASTQ. I believe IGV tool can do that. Run this FASTQ file in STRait Razor. Problem is I do not have a Linux OS machine. I understand IBM clones can run either a Windows OS or Linux OS. With dual booth up. I just started playing around with MAC OS Lion. I am new to these systems outside of Windows. Thoughts?
Oh! The other problem is only THREE complete genome of the Abusir was provided. Therefore only 3 datasets needs to be processed. The remaining mummies only their uniparental markers were released. Am I correct?
Am I talking to the wall? He! He! HE!
===
STRait Razor: A length-based forensic STR allele-calling tool for use with second generation sequencing data
Abstract Recent studies have demonstrated the capability of second generation sequencing (SGS) to provide coverage of short tandem repeats (STRs) found within the human genome. However, there are relatively few bioinformatic software packages capable of detecting these markers in the raw sequence data. The extant STR-calling tools are sophisticated, but are not always applicable to the analysis of the STR loci commonly used in forensic analyses. STRait Razor is a newly developed Perl-based software tool that runs on the Linux/Unix operating system and is designed to detect forensically-relevant STR alleles in FASTQ sequence data, based on allelic length. It is capable of analyzing STR loci with repeat motifs ranging from simple to complex without the need for extensive allelic sequence data. STRait Razor is designed to interpret both single-end and paired-end data and relies on intelligent parallel processing to reduce analysis time. Users are presented with a number of customization options, including variable mismatch detection parameters, as well as the ability to easily allow for the detection of alleles at new loci. In its current state, the software detects alleles for 44 autosomal and Y-chromosome STR loci. The study described herein demonstrates that STRait Razor is capable of detecting STR alleles in data generated by multiple library preparation methods and two Illumina® sequencing instruments, with 100% concordance. The data also reveal noteworthy concepts related to the effect of different preparation chemistries and sequencing parameters on the bioinformatic detection of STR alleles.
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
Quote: ". A separate program is currently in development that reads the colon-delimited text file(s) output by STRait Razor and detects the two allele calls with the highest count values (if any), comparing them based on a user-defined abundance ratio to make homozygous and heterozygous allelotype calls with confidence. Presently, STRait Razor does not filter reads based on FASTQ quality information. However, the flanking region detection and small-repeat match verification performed by STRait Razor can be considered inherent filtering steps. Quality based filtering may be incorporated into future versions of the program. In its current format, STRait Razor is designed to detect all the known alleles at each of the tested loci, according to the allelic information listed in STRBase and the Y-Chromosome Haplotype Reference Database (YHRD) [27,28]. These alleles are defined in the modules used by the software and may be modified by the user to include other rare or undocumented variants. In the future, the software may be altered to allow for the intuitive calling of alleles based on repeat length alone, without the need for allelic definitions.
Conclusion In its current state of development, STRait Razor offers forensic DNA analysts a simple and effective means of detecting STR alleles in SGS data. I
"
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
OH! I believe they are talking SAM Files " reads the colon-delimited text file(s"
Oh no! It works on Win also version 3.0 Quote: The short tandem repeat allele identification tool (STRait Razor), a program used to characterize the haplotypes of short tandem repeats (STRs) in massively parallel sequencing (MPS) data, was redesigned.
Written in a portable compiled language, C++, STRait Razor v3.0 functions on all major operating systems including Microsoft Windows, and it has cross-platform multithreading support.
Note that STRait Razor was designed initially for capturing short tandem repeats (STRs), but it can now detect single nucleotide polymorphisms (SNPs) and insertions/deletions (indels). The presence of these anchors in the correct orientation
STRait Razor v3.0 provides a marked improvement of the allele identification strategy employed by its previous versions. STRait Razor v3.0 is fast, in part owing to its development in a compiled programming language (C++) and in part to an indexing strategy that is tailored for fast approximate search of anchor sequences. STRait Razor v3.0 is multithreaded, allowing it to exploit multicore CPUs. Further, it no longer requires regular files and instead can use the standard input stream, allowing it to process compressed File formats and generally function in analysis pipelines. STRait Razor v3.0 also has the ability to selectively filter loci by type (e.g., one can attempt to find [b]only autosomal loci or any other user-defined type). The tool can optionally place haplotypes onto the positive strand, and low-frequency haplotypes can be excluded from the output, thus simplifying the reporting process. STRait Razor v3.0 also uses a conservative set of requirements used to resolve ambiguous haplotype identities, and it has the ability to compute the length-based nomenclature of STRs without prior expectations on haplotype lengths.
along with standard configuration files that will enable it to be used on a variety of MPS systems. Comments and findings that could improve the performance of STRait Razor v3.0 are welcome and encouraged.
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
I don't think this program is saying you can pull STRs from SNP data. If this was possible there would be no need for separate STR and SNP companies. Particularly when dealing with the Y-Chromosome.
Think of it as you trying to query specific letters (SNP) and numbers (STR) from a 10 paragraph output of text relating to the analysis of some object (Blood). The issue is the tool you used to generate those 10 paragraphs ONLY produces differing sequences of alphabetical letters. Therefore there are no Numbers in the text to query.
You would then use a SECONDARY program to produce and query any numerical data. In DNA some folks only want the letters, other folks only want the numbers. Some need both. The numbers change faster than the letters so they can tell you more information about recent events.
What is clear is when you run that program and it's says it's going to "test 100,000 to 500,000 SNPs". That is ALL ITS GOINT TO DO: Get you the values for those specific number of SNPs and NOTHING more. Or those specific STRs.
Also think of it as a sex working going to the clinic to get tested for HIV. The test is ONLY look for the HIV virus (or however it works) its not going to tell her if she has Sisyphus, or Herpes or anything else, its only designed to look for that specific things and it she wants more she will have to take additional tests to look for those other specific diseases.
Under a Y-Chromosome perspective there could be recent events that makes an STR profile look like a certain haplogroup be it the Atlantic modal halotype of European R1b, the Bantu modal STR pattern of E1b1a8 or the Cohen modal Haplotype of Jews.
In the case of Jews and the CMH, it is highly split between 2 lineages, both J1 AND J2. Its is over 80% of their CHM when you test 6-12 STRs. What about the rest? Well that STR haployype is split between the various other haplogroups including E/G/H/Q/R.
E/G/H/Q/R. Are SNPs, CHM is a 6 value STR profile. It exists within all the haplogroups listed above but mostly in J1/J2. If you test positive for CHM you would need a SNP test to know if you are Hap J or something else. Alternatively, If you test positive for J1/j2 you would need an STR test to know if you are positive for CHM because when you tested for J you ONLY tested for the presence of M172 and M267. NOTHING MORE. You dont know if you are Cohen Modal or the Arab Model.
Posts: 2463 | From: New Jersey USA | Registered: Dec 2007
| IP: Logged |
posted
@ AstenB. Appreciate your input but ...man, your analogies stink. No dis-respect intended.
I am not sure I agree. First off, the data produce are ONLY "letters" not "numbers". If it is repeated 5 times then that is 5 repeats. (eg ATTTAATTTAATTTAATTTAATTA here ATTTA is reoated 5 times. The software should be able to count the five) And keeping in mind STRs are really organized or linked SNPs that are repeated. So we are talking apples vs apples in this context. YDNA or autosomal DNA should NOT natter. The problem to overcome will be the "address/location" where the specific or STR of interest starts. That is why I asked the question above about RsXXXXX?. I assume there is a software to do that.
The question is where does the first ATTTA start on a genome that have over several million bases/alleles/SNPs?
Here is a quote from another website with people working on the project using TreeMix when it just came out. We can do our own analysis. Things are much more improved compared to when the Amarna data was released. I challenged Davidski to run TreeMix on some data to prove my point I was making. He accepted the challenged then came back a day a later and said he would not. And deleted prior comments leading up to it.
He are some excerpts from a dialogue using TreeMix(this is Dienekes)
Quote: "Zack said: 1. I ran TreeMix on the Reich et al Indian Cline dataset (alongwith CEU, Yoruba and Onge). And I found a 57% migration edge from the Onge to Yoruba. [sarcasm]Obviously, there was back-migration from India to Africa.[/sarcasm] 2. Further evidence for continent-wide Eurasian admixture in Africa that had been previously undetected due to ascertainment bias that neglected African variation 3. Using the HGDP San as outgroup there is a 63% migration edge from (Basque, Sardinian) to San
4. All joking aside, I would love to see you actually attempt this TreeMix run if you haven't already done so. Why, because sub-Saharan African cattle are by-and-large African maternally and Zebu paternally, so some group introduced Zebu cattle to Africa. 5. When you have Shorthorn cattle found in South West Ireland, North Africa and down the coast to Senegal and inland to the Central Sahel. 6. My analysis cited by Razib in this post was based on 166,770 SNPs. Following Joe Pickrell's proposal of using -k 1000 for 650K data, I have repeated the experiment with -k 250. The first migration edge added was again: (Basque,Sardinian)-to-Yoruba (64%) 7. The slight greater affinity of West Eurasians and Africans is caused by geographic proximity and some admixture events 8. So, I have hypothesized that modern humans were evolving in parts of Africa pre-100ka, especially in a "Green Sahara", slowly achieving anatomical modernity. The de-greening of the Sahara would have pushed some of them into other parts of Africa, as well as into the Near East, were we first find them c. 100ka. 9. Dienekes 5 years ago - It does seem that Yoruba, Mandenka, and Bantu are shifted relative to San in a West Eurasian direction:"
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
The point? We can do our own analysis. TreeMix software uses SNPs (and STRs)to determine migration edges. We have the raw data of the Abusir mummies. Tic! Toc!. We can download datasets for other populations from HGDP, HAPMAP, 1000genomes etc. We can plug in other Africans and run TreeMix against the Abusir and these other Africans. The Authors refused to do that in their study. We do not need "lying Europeans".....now. We can do supervised and unsupervised runs. I expect DNAConsultants are being pressured NOT to release any comparison review as they did with the Amarnas.
BTW - Keep in mind STR's are really repetitions(repeats) of a series of base pairs or alleles. SO I would think it is an easy conversion from SNP to STRs. The problem is where the first repeats starts. I have information on which of the 23chrosomes these popSTR resides. The information is out there.
Maybe when I retire this will be my serious hobby. But I am a long way off.
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
Here is the thing though - As I know it, SNP data is not testing repeats. Its doesn't retrieve that information. For Example if I go into my raw data. RIGHT NOW.....this is the exact data extracted by the Illumina sequencer Chip (Version 2 at 23andme). Back then it was 300-400k SNP's. Right now 23andme probably gives you 1 Million or more.
Lets take a look 5 random SNP's from my Chromosome 5:
1 - intergenic rs10036054 2352730 C or T C / C 2 - intergenic rs10039326 2513346 A or G A / G 3 - intergenic rs1002647 2560537 A or G A / A 4 - intergenic rs10038374 2823353 A or G A / G 5 - ADAMTS16 rs10037656 5183783 A or G A / A
You dont get the repeats. Looking at SNP number 2 I get the name of the SNP "rs10039326" and my Genotype "A/G". And you basically get this Hundreds of Thousands of times all at SPECIFIC SNP's that are known to be somewhat different when looking at world wide populations.
If you could use a program to get STR information from SNP's then companies would not need you to sumbit new cheek swabs. You could just send your 23andme Raw data over to DNA Tribes. NO instead, if you want tribes STR analysis (or ANY STR analysis) you have to swab the cheek. If you send them your 23andme Raw data they can ONLY do a SNP analysis of that data....SNP imput = SNP output.
Y-dna and Autosmal dna DO matter because If you are looking for autosomal SNPs you will not test for SNP's on the Y-chromosome because the Y nor the mtDNA contain Autosomal data.
IN THE FUTURE...yeah if there was a test that just sequenced "every damn thing" you could potentially have all the data but there are issues with this: 1 - TIME - You are going to need a super computer to crunch all that data, I hope you got a snickers cause you aint going anywhere. 2 - COSTS - It will not be cheap, it will not be 100. 3 - SIZE - You are going to end up with a Exabyte of data. 4 - We Dont even really know how large the genome IS. Nearly ever year there are resolving and "discovering" new SNP's, particularly on the Y-chromosome. IN actuality they are not "discovering" anything because those SNP's were already there, the number of positions in the human genome could reach into the quadrillions or something.
Now if you can give an alternative example using evidence that shows you can get STRs from SNPs (when we already know you cannot get SNPs from STRs) The floor is yours.
Posts: 2463 | From: New Jersey USA | Registered: Dec 2007
| IP: Logged |
posted
You can get repeats from BAM files, I'm just salty that Xyyman took all this time to look shit up and post "tic-toc" instead of actually trying to genotype STRs on his own... I even offered to give him a tutorial. I'm guessing the private companies like 23andMe call variants for you before allowing you to download the raw Data... what is the file format for 23andME??
And to hell with reconverting BAM to SAM files ...hell no... The abusir 3 are already sorted, indexed and ready to go, though I'm not gonna lie I am having trouble getting calls but I only started yesterday.
Btw Xyyman "The question is where does the first ATTTA start on a genome that have over several million bases/alleles/SNPs?" The term you're looking for is "base pairs" and that is one hell of a question from a "science guy"
If I didn't already have 23 gigabytes worth of Genetic data, I wouldn't even be doing this for you... and you're up here spamming the board with misleading info. God forbid anyone wanted to start up from here.
Edit Actually..I'm lying above, I have 23 Gigs on my internal HDD, 7x's that amount or so on an external Harddrive ...smh
Btw, the FastQ files are also up for download ...no conversion needed... tic-toc
Posts: 1781 | From: New York | Registered: Jul 2016
| IP: Logged |
posted
lol! Ok theMaster. I was trying to grasp the indexing thing. But now that you mention it. If it is indexed then determining where the repeats start should be easy. Duh! I should have picked that up. yeah. I missed that one.
One point for you!
"The question is where does the first ATTTA start on a genome that have over several million bases/alleles/SNPs?" The term you're looking for is "base pairs" and that is one hell of a question from a "science guy" "
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
Ok. I wasn't aware the FASTQ files were available(another point for you). Me being lazy and spamming. ...the board. But I will give you your props. But I am now getting into this level of details. I left it up to the experts/researchers to process the raw data. I guess I will have to start processing the data myself.
@AstenB - as you heard STRs can be obtained from these files. Keep in mind BAM files are binary SAM files are text delimited. The computer does not care or know if it is processing autosomal or uniparental markers. It is "one's and zero's". SAM tools give you the text and that is where "indexing" comes in. Essentially IDing the position of the bases/nucleotides. Again uniparental markers vs autosomal does not matter. But , You may have to "tell" the computer software what it is looking at before you do the "run and call". I am not there yet. Now getting into it. But that is the only way I can see the computer will know the difference before a run by the operator telling it it is data for uniparental markers.
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
B. No relevance Bro. See above. BAM files are 1's and 0's ie binary there is no text. SAM tools get the text from binary. That is why you need both. It only took me a few days to grasp this. I can do runs if I have the time. You have a lot to learn. But don't be mad. We are depending on you since you have the time. I don't.
@elMaestro. "basepairs" damwn! you are right. missed it again. basepair=alignment.- kicking myself!.
quote:Originally posted by beyoku:
quote:Originally posted by Elmaestro: [QB] You can get repeats from BAM files,
Do those BAM files have the 13 universal Codis markers?
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
quote:Originally posted by Elmaestro: [QB] You can get repeats from BAM files,
Do those BAM files have the 13 universal Codis markers?
Well, the Markers have to be Identified by the program you're using to Genotype. Xyyman is partially right about what the BAM files actually are. I'm about to switch over to LobSTR and see if I can get further, The Markers I found so far were weak and aren't of the popular CODIS 13... HipSTR for example tries to ID hundreds of thousands of repeats... Considering the Size of the Bam files I feel like I can get somethingPosts: 1781 | From: New York | Registered: Jul 2016
| IP: Logged |
posted
Bad news, after multiple attempts and methods I failed to get any useful STR data... Even from the fastq files. All the STR calls I got were generic. I couldn't Identify them with any program so I even checked what was going on manually (by literally looking at the loci), and it turns out their's no bps at all in the regions where we'd find these markers... so to answer Beyokus Question above no, neither do the Fastq files have em, they probably weren't even sequenced.
If anyone want a copy of the generic STR calls for whatever reason pm me, though I might warn you, you're on your own from there... I haven't bothered classifying them or researching the nomenclature.
my b Xyyman
Posts: 1781 | From: New York | Registered: Jul 2016
| IP: Logged |
So, did these "single individuals" had admixture, or was "purely" these SNPs found in these "single individuals"? Considering that most seem to carry subclades of some sort.
quote:Originally posted by the lioness,: why are you saying they are low in frequency?
Because it is, hence my previous statement.
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
quote:Originally posted by Ish Gebor: @ the lioness, how do you feel about the nazi history these institutes have? I am referring to University of Tuebingen and Max Planck.
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
quote:Originally posted by Ish Gebor: @ the lioness, how do you feel about the nazi history these institutes have? I am referring to University of Tuebingen and Max Planck.
posted
Those European researchers are slippery as a snake. What did I tell you?
They are not going to make the same mistake twice. But I am sure with what they sequenced and released information can be obtained. This is where DNATribes and other companies with the capabilities can come in useful. Lucas Martin is dead (RIP) so...someone needs to step in and step up. I don't know enough to do a deep dive.
What About EDAR, Duffy gene, sickle cell, hypertension gene and other genes with high frequency in Africans?
"they probably weren't even sequenced"
quote:Originally posted by Elmaestro: [Q] Bad news, ........, they probably weren't even sequenced.
...
my b Xyyman [/QB]
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
So it seems like the FULL genomes(3) were not really sequenced and released?! May be not released but sequenced. I thought about that last weekend. If they made the STR data available that would be a major screw up on their part.
I thought they would REMOVE that sequence(STR) data before they released the rest of the genome.
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
Looks like they were removed from the BAM file before being released or they weren't sequenced. My money is they were removed.
As I said it is impossible for the Amarnas who were SSA to be any different from the Abusir which is a few hundred miles away ...and before the age of intercontinental travel.
trust me...it was removed. That is how these "lying Europeans" operate.
either "data not shown", "through private communication". or "selective sampling".
I know all their games and tactics. It is getting like a broken record
quote:Originally posted by beyoku:
quote:Originally posted by Elmaestro: [QB] You can get repeats from BAM files,
Do those BAM files have the 13 universal Codis markers?
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
I just had an idea. Anyone has the BAM files for the Amarnas? were they released as the Abusir mummies?
----
BAM is the compressed binary version of the Sequence Alignment/Map (SAM) format, a compact and index-able representation of nucleotide sequence alignments. Many next-generation sequencing and analysis tools work with SAM/BAM. For custom track display, the main advantage of indexed BAM over PSL and other human-readable alignment formats is that only the portions of the files needed to display a particular region are transferred to UCSC. This makes it possible to display alignments from files that are so large that the connection to UCSC would time out when attempting to upload the whole file to UCSC. Both the BAM file and its associated index file remain on your web-accessible server (http, https, or ftp), not on the UCSC server. UCSC temporarily caches the accessed portions of the files to speed up interactive display.
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
To those who are interested. Binary files are essentially 1's and 0's. Remember elementary or High School Math depending on your educational background? Computers process in 1's and 0's but human's read or process through alpha/numeric characters. That is why SAM Tools is needed to convert BAM files which are in binary. ie 1's and 0's to ATTTC etc The nucleotide code or bases. The problem is you end up with a large string of AAATTC etc. Comes like a long street in NY City. They need to be index or numbered to make sense and to help with location. Where do city blocks begin and end. That is why indexing/numbering is so important.
After indexing the genes or SNP or STR can be located. Eg if you have a city clock with 1000 houses and all are only four colors the numbering the houses will help in "location or addressing"
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
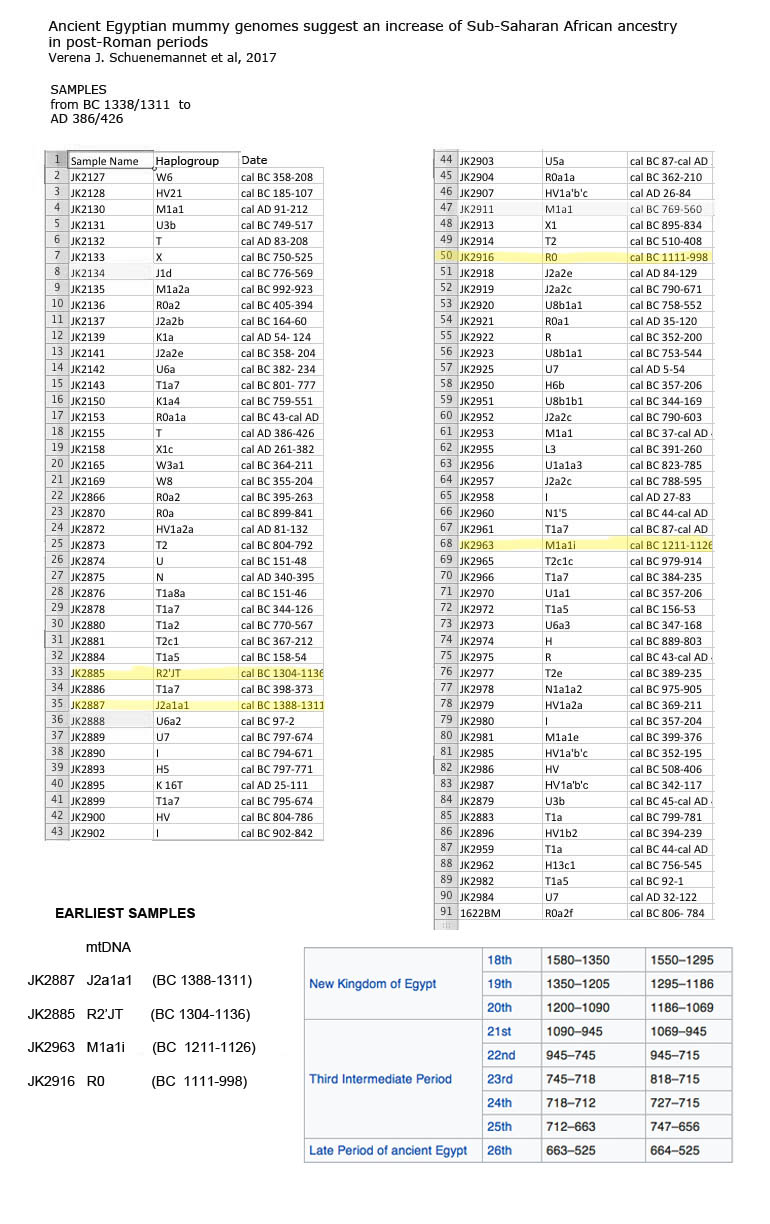
For the newbies. From The JAMA Report. These were SSA Africans from the New Kingdom?
---- An up to 30-fold testing of polymorphic autosomal microsatellite loci via the combined use of the Identifiler and AmpF\STR Minifiler kits (Applied Biosystems) yielded complete data sets for all 8 markers in 7 mummies (Thuya, Yuya, Amenhotep III, Tutankhamun, KV55, and both female mummies from KV35) but only partial data for both KV62 fetuses and the KV21A and KV21B mummies (Figure 1). Repeated attempts to complete the profiles in the 4 latter mummies were not successful; however, we were able to replicate some of the results for the previous mummies more than 4 times in the second, independent laboratory (Figure 1). Moreover, because these profiles differed from those of the laboratory staff and were not identical to the ones established for the control group, the data were considered authentic.
Conclusion Using a multidisciplinary scientific approach, we showed the feasibility of gathering data on Pharaonic kinship and diseases and speculated about individual causes of death.
The 18th dynasty (circa 1550-1295 BC) of the New Kingdom (circa 1550-1070 BC) was one of the most powerful royal houses of ancient Egypt. The pharaoh Akhenaten, who ruled from circa 1351 to 1334 BC, is considered one of the most controversial of the Egyptian pharaohs, because his attempt to radically transform traditional religion affected all facets of society and caused great turmoil.
Author Contributions: Drs Hawass, Gad, Zink, and Pusch had full access to all of the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
Study concept and design: Hawass, Gad, Zink, Pusch.
Analysis and interpretation of data: Hawass, Gad, Ismail, Khairat, Fathalla, Hasan, Ball, Wasef, Fateen, Amer, Gostner, Selim, Zink, Pusch.
Drafting of the manuscript: Hawass, Gad, Zink, Pusch. ----
And now from The Abusir mummies - QUOTE" ll sampled remains derive from this community in Middle Egypt and have been radiocarbon dated to the late New Kingdom to the Roman Period [cal. 1388BCE426CE, Supplementary Data 1}. In particular, we seek to determine if the inhabitants of this settlement were affected at the genetic level by foreign conquest and domination, especially during the Ptolemaic [33230BCE} and Roman [30BCE395CE} Periods
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
posted
At this point in time a report like this should be multi-disciplinary and have multiple forms of supporting data. On the genetics side all the DNA haplogroups should be listed, make and female. On the physical anthropology side, all the mummies identified, even if only by reference number, since they came from a museum collection. That would allow for other folks to cross check and do further analysis. Otherwise, it is just another half baked study that can be used to bolster a Eurocentric agenda.
Posts: 8889 | Registered: May 2005
| IP: Logged |
So, did these "single individuals" had admixture, or was "purely" these SNPs found in these "single individuals"? Considering that most seem to carry subclades of some sort.
quote:Originally posted by the lioness,: why are you saying they are low in frequency?
Because it is, hence my previous statement.
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
quote:Originally posted by xyyman: 9 post in 12years!! Last post was in 2005. You waited 12years to write.."stop" wtf you wierdos come from? Suck puppet account by the owner?
@Lioness - you don't have to read my post. And i do read Ish's. Very good researcher.
So how about it d intellect. Any thing to contribute to my suggestion. Can you identify African genes based upon the released BAM files?
quote:Originally posted by d-intellect: Stop.
I was trying to help you as someone who doesn't feel this study is the end all/be all that the mainstream is making it out to be. As a casual observer to this board, your posts to yourself have become counterproductive from an outsider's perspective. It actually makes you look like the "weirdo" having a difficult time countering the findings of the project outside of the friendly confines of this board. I won't pretend to be a geneticist so I will remain in observation mode. Carry on this way if you must.
Posts: 10 | Registered: Jun 2005
| IP: Logged |
posted
Northeast African genomic variation shaped by the continuity of indigenous groups and Eurasian migrations - Nina Hollfelder
Eurasians migrated 700AD
AEians cannot but be anything else but indigenous Africans. I am 110% sure. There is no doubt. I said this from the git go.
quote:Originally posted by the lioness,: [Q]
quote:Originally posted by xyyman:
1. It is impossible for the Abusir mummies to be anything but Africans.
why? [/Q]
Reading the summary Elmaestro. It is not what you think. I myself got caught in the sensationalism title without reading. 1. It is supporting the fact the Ancient Egyptians are Sudanese sand they occupied regions along the Nile BEFORE the Arab conquest about 700AD.
Is this a rebuttal to Abusir paper? Lol!
Man you people are like whores. SMH
----- Authors summary This admixture process largely coincides with the time of the Arab conquest, spreading in a southbound direction along the Nile and the Blue Nile. Nilotic populations occupying the region around the White Nile show long-term continuity, genetic isolation and genetic links to ancestral East African people. Compared to current times, groups that are ancestral to the current day Nilotes likely inhabited a larger area of northeast Africa prior to the migration from the Middle East as their ancestry component can still be found in a large area. Our findings reveal the genetic history of Sudanese and South Sudanese people, broaden our knowledge on demographic history of humans, and quantify the impact of large-scale historic migration events in northeast Africa.
t disputes Abusir.
also Quote: The Nilotes are predominantly pastoralist populations, they live in Uganda, Ethiopia, Kenya, Tanzania, and are the most ****PROMINENT*** ethnicity in South Sudan. They are traditionally strongly endogamic which could account for low levels of admixture. In terms of specific Nilotic populations, the f3 test showed no significant signal of gene flow with external populations for the Nuer and Baria (Fig 3A), however, we detected indications of external
-------------------- Without data you are just another person with an opinion - Deming Posts: 12143 | From: When you have eliminated the impossible, whatever remains, however improbable | Registered: Jun 2007
| IP: Logged |
quote:Originally posted by xyyman: [QB] Northeast African genomic variation shaped by the continuity of indigenous groups and Eurasian migrations - Nina Hollfelder
Eurasians migrated 700AD
AEians cannot but be anything else but indigenous Africans. I am 110% sure. There is no doubt. I said this from the git go.
quote:Originally posted by the lioness,: [Q]
quote:Originally posted by xyyman:
1. It is impossible for the Abusir mummies to be anything but Africans.
why? [/Q]
Reading the summary Elmaestro. It is not what you think. I myself got caught in the sensationalism title without reading. 1. It is supporting the fact the Ancient Egyptians are Sudanese sand they occupied regions along the Nile BEFORE the Arab conquest about 700AD.
yeah but you said there was no Arab conquest
Posts: 42918 | From: , | Registered: Jan 2010
| IP: Logged |
quote:Originally posted by the lioness,: xyyman, where did haplogroup J2 originate?
and where is the greatest diversity of J1?
What is the genetic coding difference of the gene expresion markup between these Haplo types? And what has led to this "possible" genetic coding differences?
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
quote:Originally posted by xyyman: [QB] Northeast African genomic variation shaped by the continuity of indigenous groups and Eurasian migrations - Nina Hollfelder
Eurasians migrated 700AD
AEians cannot but be anything else but indigenous Africans. I am 110% sure. There is no doubt. I said this from the git go.
quote:Originally posted by the lioness,: [Q]
quote:Originally posted by xyyman:
1. It is impossible for the Abusir mummies to be anything but Africans.
why? [/Q]
Reading the summary Elmaestro. It is not what you think. I myself got caught in the sensationalism title without reading. 1. It is supporting the fact the Ancient Egyptians are Sudanese sand they occupied regions along the Nile BEFORE the Arab conquest about 700AD.
yeah but you said there was no Arab conquest
quote:African and Middle Eastern populations shared the greatest number of alleles absent from all other populations (fig. S6B).
Sarah A. Tishkoff et al. The Genetic Structure and History of Africans and African Americans
Posts: 22234 | From: האם אינכם כילדי הכרית אלי בני ישראל | Registered: Nov 2010
| IP: Logged |
posted
I wouldn't bet on accuracy of ancient African mummies decayed DNA results, because a comprehensive, functional analysis of decaying mRNA transcripts has not been performed until now. Beside,I am suspicious of DNA results from European and American labs, because of different tests conclusions from the same subjects done in their labs.
I have no doubts that many DNA tests results done by them are influenced by their colonial racial, territorial and economic ambitions. Oh...one of their most ridiculous deduction I saw so far, was a facial painting of a pre-historic blue eyes European male, which was done based on one found tooth's DNA. If they are capable of such corruption, image what they may do to ancient African DNA results.
-------------------- ---lnnnnn* Posts: 198 | From: USA | Registered: Aug 2014
| IP: Logged |
posted
I understand that the paternal genetic profile of the Natufians was African, so would that not make the Abusir mummies more than just 6-15% African?
Posts: 1568 | From: Pluto | Registered: Sep 2008
| IP: Logged |
quote:Originally posted by sudaniya: I understand that the paternal genetic profile of the Natufians was African, so would that not make the Abusir mummies more than just 6-15% African?
Lazaridis et al. (2016) tested the first ancient DNA samples from the Mesolithic Natufian culture in Israel, possibly the world's oldest sedentary community, and found that the male individuals belonged either to haplogroups CT or E1b1 (including two E1b1b1b2 samples). These are to date the oldest known E1b1b individuals.
-------------------- a sense of National sovereignty is the heart of imperialism
Posts: 42918 | From: , | Registered: Jan 2010
| IP: Logged |
quote:Originally posted by Elite Diasporan: I have a question. Were any crania/limb proportion works done on these mummies?
i googled a bit and couldn't find anything. could try emailing the University of Tubingen i guess.
Posts: 660 | From: Canada | Registered: Mar 2017
| IP: Logged |




 UBBFriend: Email this page to someone!
UBBFriend: Email this page to someone!




![[Smile]](smile.gif)

![[Wink]](wink.gif) ...no conversion needed... tic-toc
...no conversion needed... tic-toc


![[Big Grin]](biggrin.gif)


 Printer-friendly view of this topic
Printer-friendly view of this topic